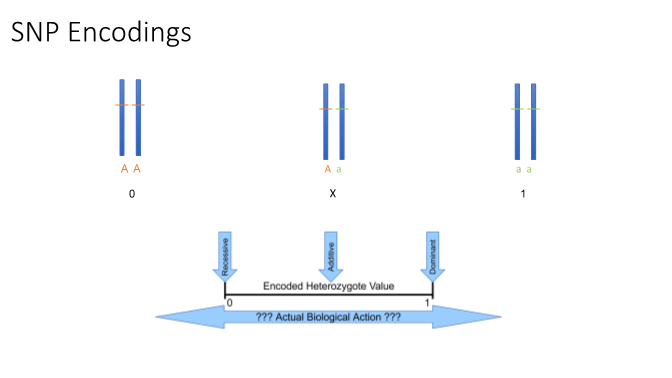
Choosing between traditional methods for encoding single nucleotide polymorphisms (SNPs) in association studies, including additive, dominant, recessive, and codominant, requires making an assumption about the manner in which the coded risk allele acts. In accordance with Mendel’s patterns of inheritance, given referent allele, A, and alternate (or coded risk) allele, a, all encodings assume that the AA (homozygous referent) genotype incurs no risk and aa (homozygous alternate) genotype bears full risk (Figure 1). The assumed heterozygous (Aa) risk, however, varies according to the chosen encoding method. For each encoding, the assumed risk accrued by one copy of the alternate allele (Aa) in relation to two copies (aa) varies: Aa is coded to bear 0%, 50%, or 100% the risk of aa for recessive, additive, and dominant encodings, respectively. However, heterozygous risk could, in actuality, lie anywhere between 0% to 100% of the risk of a homozygous alternate genotype. Additionally, choosing only one of these encodings is restrictive, as SNPs across the genome are unlikely to demonstrate identical genetic models. Testing all encodings increases the computational and multiple testing burden, thereby limiting the ability to identify true signals. This issue becomes more complicated when dealing with epistasis (genetic interactions): testing all combinations of encodings for every SNP in a SNP-SNP interaction pair is a daunting task with far too many tests for which to adjust.
We developed an alternative to the current paradigm: the elastic data-driven genetic encoding (EDGE), a method to flexibly assign each SNP with a unique heterozygous encoding prior to interaction analysis (Hall et al., PLoS Genetics (accepted)). With simulations, we demonstrate that this novel method is more accurate and robust than traditional encoding methods in estimating heterozygous genotype values, reducing the type I error, and detecting SNP-SNP interactions. We further applied the traditional encodings and EDGE to biomedical data from the Electronic Medical Records and Genomics (eMERGE) Network for five phenotypes, and EDGE identified a novel interaction for age-related cataract not detected by traditional methods, which replicated in data from the UK Biobank[1]. EDGE provides an alternative approach to understanding and modeling diverse SNP actions and is recommended for studying complex genetics in common human phenotypes.
References
Relevant Links